
Prefiltering
This tool does the following tasks:
- Rename the FASTQ reads to serial numbers (eliminate spaces in the sequence name)
- Filter out too short reads
- Truncate the reads at the low-quality nucleotide if necessary
- Remove the adapter sequences
This tool runs under Windows platform and requires .NET framework 4
How to use:
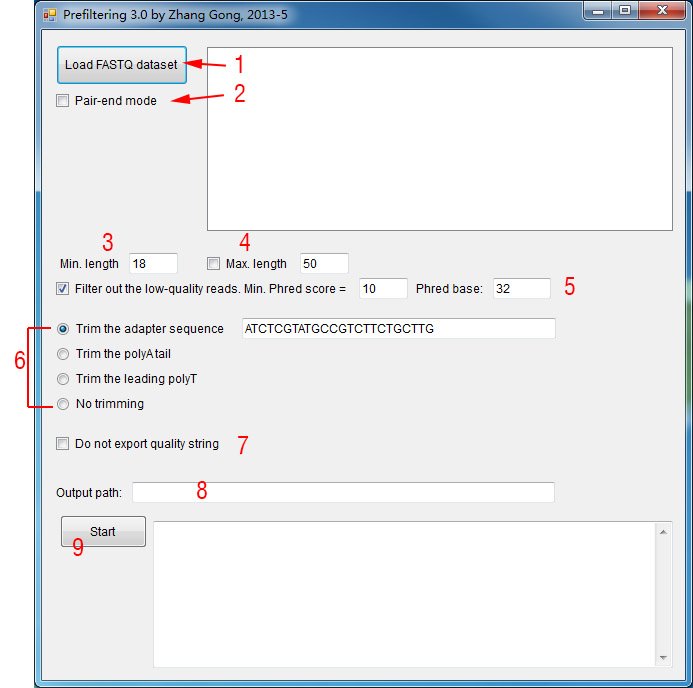
1. Click this button to load FASTQ dataset(s). You can load multiple FASTQ datasets at the same time and the program will process them one by one.
2. If your FASTQ files are paired for pair-end sequencing, check this box to make it as pair-end mode. In this mode, if one end is filtered out, the other end is also discarded.
3. Set the minimal length. Note that FANSe2 requires at least 14 nt reads.
4. If checked, all reads that are longer than the specified value will be truncated after the set value of nucleotides.
5. If checked, the program performs a quality check. Nucleotides with min. phred score will be considered as high quality nucleotides. Reads will be truncated at the first low quality nucleotide. After the truncation, the read will be discarded if it is shorter than the min. length setting.
6. Adapter trimming mode.
7. Do not export the quality string. This option is useful for FANSe2 since FANSe2 do not need the quality information. This will largely reduce the FASTQ file size. However, if you are planning to use the filtered FASTQ file in other programs, please do not check this box.
8. Specify the output path. If not specified, the output FASTQ file will be in the same folder as the input file.
9. When all the above has been properly set, click this button to start prefiltering.
The output files will be added "-f" in their file name. For example, if you prefilter a file "ABC.fastq", the output file will be named as "ABC-f.fastq".