
GeneQuantRefSeq
This tool is an RNA-seq quantification tool.
You map RNA-seq reads to RefSeq-RNA reference sequences using FANSe/FANSe2. Then, you can use this tool to count the read count for each gene and its RPKM value to estimate its expression level (Mortazavi et al., 2008). The results are given in two lists: one list with all the splice variants separated, and another list merged all the splice variants.
This tool runs under Windows platform and requires .NET framework 4.
How to use:
Before using this tool, you should map your RNA-seq dataset to a RefSeq-RNA reference sequence set of a certain species using FANSe2. You may download these kind of datasets from UCSC or NCBI. For example, the RefSeq-RNA reference sequence set for human hg19/GRCh37 can be downloaded here. Note that many RefSeq-RNA contain the FASTA sequences with the names containing spaces. You should keep only the NM/NR ids for the name. For example, the RefSeq-RNA for mouse mm10 looks like this:
>NR_037984 1
tgaagtggctgtaagcaagagggacaattaccacaccctatctccccttc
gattccacctttgtgataacaaaattaccacagggcaggaggagttggtc
ccctaaacaggaccatctcaaacccagcttcactactgagaagctggccc
tacgccttcctcaagaggaaacacctgagcccctatccacggcatgcagg
...
Note that there is a suffix " 1" after the real NR id "NR_037984". You have to remove the suffix " 1" before mapping, otherwise there will be an error. You may use our utility "RenameRefSeq" to do this. The useable reference sequence should look like this:
>NR_037984
tgaagtggctgtaagcaagagggacaattaccacaccctatctccccttc
gattccacctttgtgataacaaaattaccacagggcaggaggagttggtc
ccctaaacaggaccatctcaaacccagcttcactactgagaagctggccc
tacgccttcctcaagaggaaacacctgagcccctatccacggcatgcagg
...
For more information, please refere to the FAQ section.
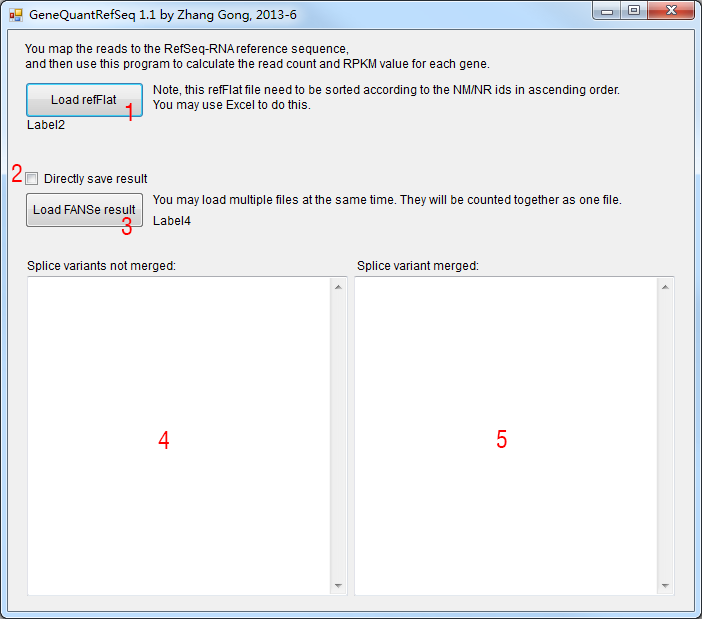
1. Load refFlat file. The refFlat file records all the information of RefSeq RNA, including the protein-coding mRNA (NM_*) and non-coding RNA (NR_*). You may download this kind of file from UCSC annotation database. But be careful, you need to sort this refFlat file according to the NM/NR ids in the ascending order. You may do this using Excel or WPS Spreadsheet or similar softwares.
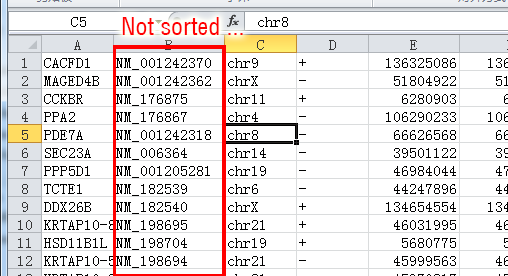
When you open the downloaded refFlat file in Excel you may see this:
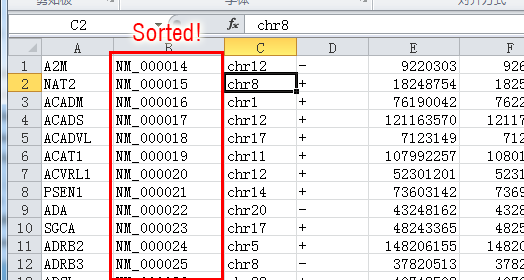
After sorting according to the column B in ascending order, the sorted table are like this:
Then you save this again back to text file format (not .xls or .xlsx format!). This is the "sorted refFlat" file that can be used in the GeneQuantRefSeq program.
2. If you want to direcly save the result into files, please tick this box. Two text files will be saved, see below.
3. Click this button to load FANSe/FANSe2 mapping result file. You can load several fanse format result files and they will be treated together as one entire file. The calculation will begin and finally export the expression list. Actually two lists will be exported as results: one list with all the splice variants separated, and another list merged all the splice variants according to the gene name. Both lists look like this:
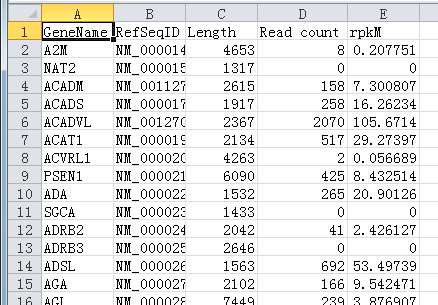
Now you get your result for mRNA quantification. Read count can be used in edgeR/DEseq for differential gene expression estimation. rpkM can be used to compare the expression level between different genes in one sample.