FANSe-CG (CorrectGenome)
![]() Download FANSe-CG 1.0
Download FANSe-CG 1.0
This tool uses the mapped reads to correct genome sequence of prokaryotes (do not support multiple chromosomes).
Sorry that the download is temporarily unavailable. The previous version needs to run 8 programs for one round of genome correction, therefore it was complained by many users. To make it more user-friendly, we are working on a easier-to-use version.
![]() This tool runs under Windows and do NOT need .NET framework.
This tool runs under Windows and do NOT need .NET framework.
![]() Citation:
Citation:
Wu X, Xu L, Gu W, Xu Q, He QY, Sun XS, Zhang G, Iterative Genome Correction Largely Improves Proteomic Analysis of Non-model Organisms. J Proteome Res, 2014 May 19.
What does it do:
- Sequence your sample, DNA-seq or RNA-seq are both OK.
- Map the reads to the reference genome sequence with FANSe/FANSe2.
- Run this tool to identify single nucleotide variations (SNVs, including substitutions and indels).
- Exports the list of SNVs and corrected genome sequence.
SNV identification:
The SNV identification in this tool is based on Fisher's Exact test on each position. This provides reproducible and robust SNV identifications. It requires
- the read coverage at this nucleotide was more than 5;
- the occurrence of predominant nucleotide is significantly more than half of the coverage (Fisher’s exact test, p<0.05 against the null hypothesis that the occurrence of the predominant nucleotide equals the half of the coverage);
- the predominant nucleotide is different from the one in the reference sequence.
For details please also refer to our published papers:
Zhong, J.; Cui, Y.; Guo, J.; Chen, Z.; Yang, L.; He, Q. Y.; Zhang, G.; Wang, T., Resolving Chromosome-Centric Human Proteome with Translating mRNA Analysis: A Strategic Demonstration. J Proteome Res 2013
Chinese Human Chromosome Proteome, C.; Chang, C.; Li, L.; Zhang, C.; Wu, S.; Guo, K.; Zi, J.; Chen, Z.; Jiang, J.; Ma, J.; Yu, Q.; Fan, F.; Qin, P.; Han, M.; Su, N.; Chen, T.; Wang, K.; Zhai, L.; Zhang, T.; Ying, W.; Xu, Z.; Zhang, Y.; Liu, Y.; Liu, X.; Zhong, F.; Shen, H.; Wang, Q.; Hou, G.; Zhao, H.; Li, G.; Liu, S.; Gu, W.; Wang, G.; Wang, T.; Zhang, G.; Qian, X.; Li, N.; He, Q. Y.; Lin, L.; Yang, P.; Zhu, Y.; He, F.; Xu, P., Systematic Analyses of the Transcriptome, Translatome, and Proteome Provide a Global View and Potential Strategy for the C-HPP. J Proteome Res 2013
The indel detection is also similar.
Experimental validation
The results in our case of Bacillus pumilus genome correction was 100% validated by Sanger sequencing. In this validation we sequenced 1994 nucleotides. FANSe2-CG gives corrected genome sequence 100% identical to the real one, no false positives and no false negatives.
Wu X, Xu L, Gu W, Xu Q, He QY, Sun XS, Zhang G, Iterative Genome Correction Largely Improves Proteomic Analysis of Non-model Organisms. J Proteome Res, 2014 May 19.
Typical applications:
- Identify SNVs (substitutions and indels)
- Generate sample-specific corrected genome sequence from DNA-seq or RNA-seq data
- Generate sample-specific corrected reference proteome sequences from the corrected genome sequence for mass spectrometry identifications
- You can perform iterative corrections using one sequencing dataset to correct 5~20% deviations.
Performance:
CorrectGenome can correct 19.2% deviations from the reference genome sequence. The corrections are fully validated by Sanger sequencing.
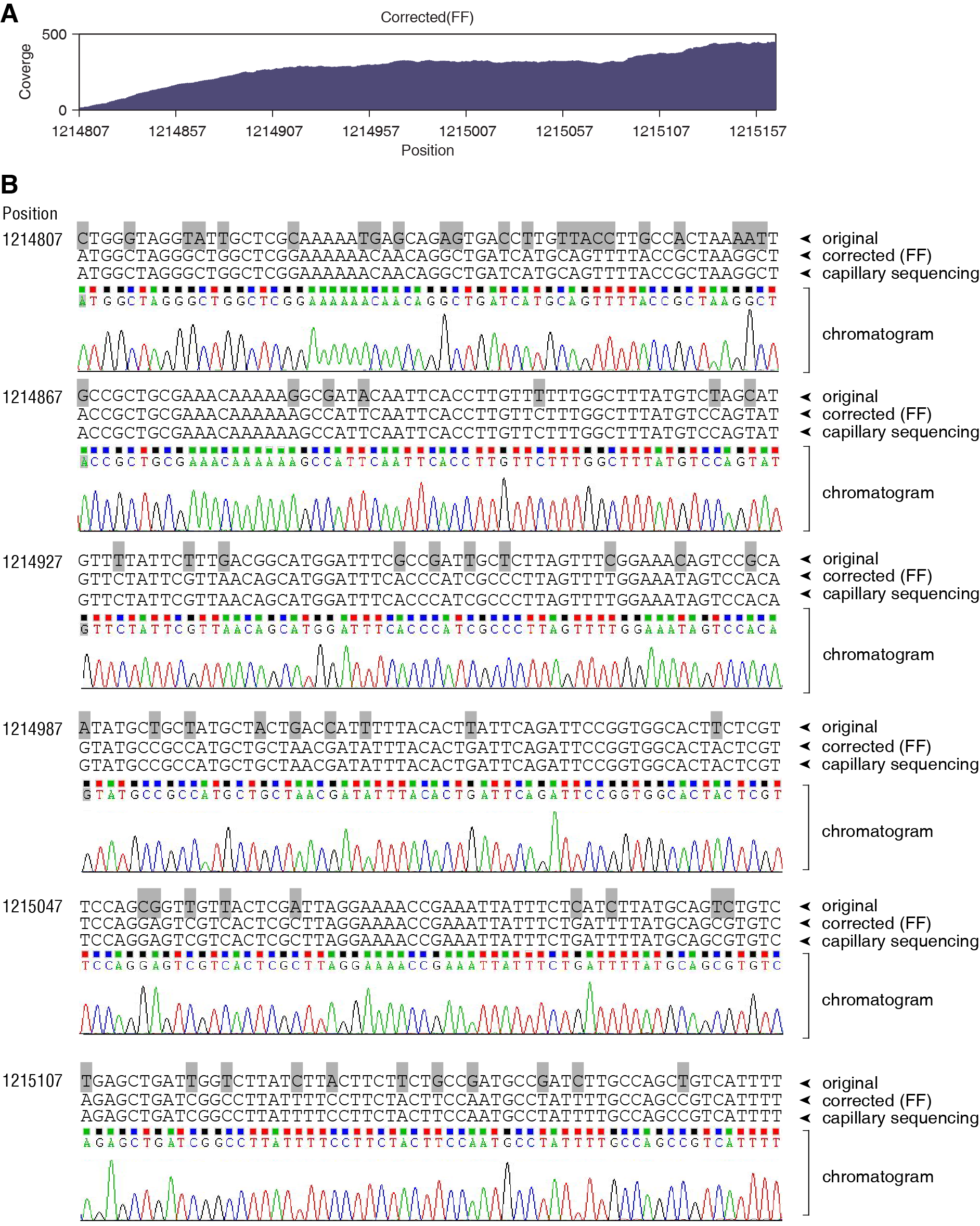
We isolated an environmental bacterial strain and determined its 16S rDNA sequence that fully matches the Bacillus pumilus SAFR-032. We performed the whole genome sequencing using Illumina HiSeq-2000 sequencer, obtaining 12.70 million pair-end reads. We iteratively mapped these reads to the Bacillus pumilus SAFR-032 genome and corrected the genome sequence for 7 rounds. In total, 182620 SNVs were identified. To validate the result of genomic sequence correction, we amplified a fragment from our B. pumilus genome and sequenced using traditional capillary electrophoresis sequencing system. This 360-nt fragment (position 1214807 to 1215166) contains 69 nucleotides that were corrected during the genome correction process, i.e. the variation rate is 19.2%. The sequencing depth of this fragment ranges from 14 to 445 (average 284.0), showing a good coverage and confident correction (A). Capillary sequencing result fully confirmed our corrected sequence, and the chromatograms showed all clear peaks (B). This showed that the iterative correction method is powerful enough to handle the fragment sequences with such high variation rate. In contrast, Bowtie2 did not map any read on this fragment, showing its incompetence in read alignment.
Figure legend: (A) The read coverage of the genomic fragment 1214807 to 1215166 (360 nucleotides) using FANSe algorithm. Bowtie2 did not map any read to this fragment. (B) Capillary sequencing validation of this region. The original reference genome (B. pumilus SAFR-032, “original”), the corrected genome sequence using FANSe + Fisher’s Exact Test (“corrected (FF)”), the capillary sequencing result and the chromatogram are aligned. 69 corrected nucleotides were marked using grey background..